医疗器械临床评价技术指导原则(2021年药监局通告第73号-附件1)
论证申报产品与对比器械广泛相似性的考虑事项
论证申报产品与对比器械是否具有广泛相似性时可能的考虑事项举例如下。需提供总结性文档论述下列因素如何支持可比性论证。某些情况下,可能需开展额外的测试以确立可比性的程度。
一、适用范围:
适应证,包括产品预防、诊断、缓解、治疗或者监护的疾病或症状
疾病的严重程度和阶段
患者人群(如年龄、性别、生理学信息)
适用部位(人体部位、器官、产品接触的组织或体液)
接触类型(如黏膜接触/侵入/植入)
与人体接触的时间
使用环境(如医疗机构、家用)
预期使用者(如由专业医务人员/非专业人士)
重复使用,包括重复使用次数或持续时间
二、技术特征
设计(如尺寸和公差;各组件如何组合使用)
材料(如化学配方、添加剂、加工方法(如铸造)、状态(如结晶状态))
技术参数和特性,如理化特性(如能量强度和类型)、波长、孔隙率、粒径、黏度、纳米技术、比质量、原子夹杂(如氮碳共渗)、氧化性、抗拉强度和降解特征等
关键性能要求
工作原理
三、生物学特性
降解性能
生物学反应(如炎性反应,免疫反应,组织整合等)
文献检索报告的参考格式
一、产品名称及型号规格
二、文献检索范围(与临床评价范围一致)
(一)方法
1.检索日期
2.文献检索人员姓名
3.文献检索覆盖的时间范围
4.文献来源及选择理由
(1)科学数据库–如中国期刊全文数据库、美国《医学索引》(MEDLINE)、荷兰《医学文摘》(EMBASE)
(2)系统综述数据库(如科克伦系统评价数据库(Cochrane Database of Systematic Reviews))
(3)临床试验注册中心(如科克伦临床对照试验中心注册数据库(CENTRAL))
包括来源数据库的选择理由,说明提高检出率的辅助策略 (如检查文献的参考书目、人工检索文献等)
5.检索详细信息
(1)检索词(关键词、索引词)及其关系
(2)所用媒体,如线上、CD-ROM(包括发布日期和版本)
6.文献选择标准
(二)结果
1.每个数据库中检索到的文献列表
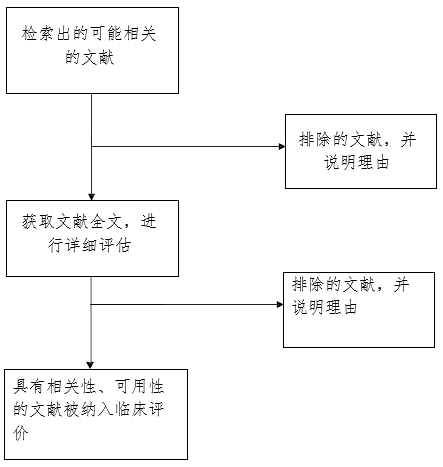
2.文献选择过程(选择方法参见附件3)
文献检索报告中文献选择方法
境外临床数据可用性的考虑事项
按照适用的临床试验质量管理规范(GCP)以及伦理要求开展临床试验时,产生的临床数据需考虑予以接受。临床数据的适用性取决于监管要求、内在和外在因素的差异性。
一、法规要求的差异
临床试验需遵守其所在监管区域的相关法规(如GCP),需考虑到产品拟上市区域临床试验质量管理规范的适用要求。临床试验未能满足上述区域适用要求的部分,需予以解释和论证。
二、内在或外在因素
临床数据适用性相关的内在和外在因素包括:
内在因素:人类遗传学特征或人口学因素,包括种族、年龄、性别等方面。
外在因素:临床实践、社会环境、自然环境、文化因素、生活行为因素、罕见病或地方性疾病等。
需采取适当方法,对可能显著影响临床数据的因素进行控制,以减少变异性。如存在剩余的变异性,需进行合理论证。在某些情况下,可能需要额外的临床数据。
建立数据评价标准的考虑因素
下列问题旨在帮助形成不同类型数据的数据评估标准,其并未完全涵盖所有试验类型或者可能的问题。
一、随机对照试验
受试者随机分入试验组或对照组,使用试验器械或对照器械(或其他干预措施),以比较试验组或对照组之间的结果和事件发生率。
有助于形成数据评估标准的问题包括:
1.是否规定入选及排除标准?
2.对照是什么?
3.分组是否真正随机?
4.是否进行了分配隐藏?
5.是否对预后风险因素的组间分布进行了充分说明?
6.上述因素的基线水平在组间是否具有可比性?
7.是否对结果评估者设盲?
8.是否对研究者设盲?
9.是否对受试者设盲?
10.是否所有随机分配的受试者都包括在分析之中?
11.是否对主要结果报告了点估计和变异指标?
二、队列研究
从使用以及未使用产品的各组中获取数据并对结果进行比较。
有助于形成数据评估标准的问题包括:
1.受试者的选择是前瞻性的还是回顾性的?
2.是否明确了干预措施?
3.是否对受试者如何分组进行了充分说明?
4.是否对预后风险因素的组间分布进行了充分说明?
5.上述因素在组间是否具有可比性?
6.是否在试验设计或者分析中,对可能的混杂因素进行了充分的控制?
7.对结果的测量是否无偏倚?
8.随访时间是否足以观察试验结果?
9.随访的比例是多少?是否有数据从分析中被排除?
10.各组间退出率及退出原因是否相似?
三、病例对照研究
选择发生/未发生规定结果的研究对象,获取其是否使用产品的信息并进行比较。
有助于形成数据评估标准的问题包括:
1.是否对如何定义受试者以及如何分组进行了充分说明?
2.病例组的疾病状态是否经过可靠评估与确认?
3.对照组是否从源对象人群中随机选择?
4.是否对预后风险因素的组间分布进行了充分说明?
5.上述因素在组间是否具有可比性?
6.在试验设计或者分析中,是否对可能的混杂因素进行了充分的控制?
7.是否在盲态下以相同方式对两组的干预措施进行评估?
8.如何定义应答率?
9.两组间无应答率及其原因是否相同?
10.是否使用了适当的统计分析?
11.是否由于干预相关因素在病例和对照间的过度匹配而影响分析?
四、病例系列研究
产品用于一系列患者并报告了结果,未设立对照组。
有助于形成数据评估标准的问题包括:
1.病例系列是否为相关人群的代表性样本?
2.是否明确了入选及排除标准?
3.所有受试者是否在相似的疾病病程进入研究?
4.随访时间是否足以对重要事件进行观察?
5.是否对使用的技术进行了充分说明?
6.是否使用客观标准或在盲态下对结果进行评估?
7.如进行子系列间比较,是否对系列以及预后风险因素的分布进行了充分说明?
参考评估方法
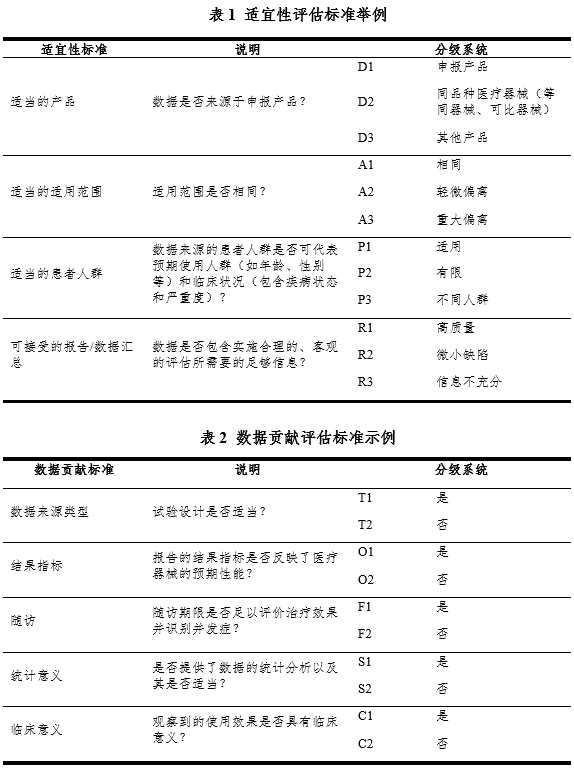
很多方法可用于临床数据的评估及权重分配。表1、表2中给出了可能使用的评估标准举例,可按顺序使用这些标准,并为适用的数据集分配权重。表1列明的数据适宜性标准虽然适用于所有医疗器械,但实际使用的方法将根据产品的不同而有所不同。
临床评价人员需根据数据来源类型对数据集进行分类,系统考虑最有可能影响结果解读的因素(表格2)。临床评价人员需在一定范围内确定哪一类问题对产品特征、研发历史以及预期临床使用更为重要。以下举例中使用的标准聚焦于较高风险产品可能关注的问题,如产品特征、结果评价方法、随访时间和完成情况以及结果的统计和临床意义等。
以下示例中,使用分配权重的方法来评估数据集对证明产品安全性、临床性能和/或有效性的贡献。当某一数据集拥有的一级分级越多时,其提供的证据的权重就越大,但并不建议将各类情形的相对权重相加构成总分。